

Chef d'équipe : Anne Joutel
Membre d'équipe : Shiraz DIB | Valérie Domenga-Denier | Virginie Dubosclard | Nicolas Dupré | Julien Guemri | Florian Gueniot | Sherlina Salignac
 |
Les maladies des petits vaisseaux cérébraux (SVD) sont responsables d’environ 25% des accidents vasculaires cérébraux ischémiques, 90% des hémorragies intracérébrales et sont une cause majeure de déclin cognitif et de handicap chez l’adulte. Notre compréhension limitée de la pathogenèse de ces maladies constitue un obstacle majeur au développement de thérapeutique. Notre équipe étudie les mécanismes physiopathogéniques des maladies des petits vaisseaux cérébraux. Nous utilisons comme modèles expérimentaux des formes monogéniques de SVD dont la symptomatologie est très proche des formes sporadiques, en particulier, la maladie CADASIL, une forme ischémique de SVD causée par des mutations très stéréotypées du récepteur Notch3, et la maladie COL4A1, une forme hémorragique de SVD causée par des mutations très stéréotypées dans la chaine alpha1 du collagène de type IV. Au cours de ces dernières années, notre équipe a généré et caractérisé plusieurs modèles souris de la maladie CADASIL et COL4A1. Dans la maladie CADASIL, nous avons établi que l'agrégation / accumulation du domaine extracellulaire du récepteur Notch3 dans les vaisseaux cérébraux est un événement central, favorisant le recrutement anormal de protéines de la matrice extracellulaire à l’origine d’une toxicité multifactorielle. Nous avons identifié un nouveau mécanisme, connectant une altération pathologique de la matrice extracellulaire microvasculaire et une surexpression des canaux potassiques voltage-dépendant, qui sous-tend la dysfonction des vaisseaux cérébraux. Enfin, nous avons montré l'efficacité préclinique de l'immunisation passive ciblant Notch3 dans une étude preuve de concept. Le but de l’équipe est maintenant de mieux comprendre le mécanisme des infarctus cérébraux, d’identifier quelles espèces de Notch3 sont toxiques, et de poursuivre l’étude du potentiel thérapeutique de l'immunisation passive contre Notch3. Dans la maladie COL4A1, nous avons récemment identifié les mécanismes cellulaires à l’origine des hémorragies cérébrales. Nous cherchons maintenant à en déchiffrer les mécanismes moléculaires. A terme, nous avons l'ambition de passer à l'étude des formes sporadiques de SVD, en commençant par l’étude de la matrice extracellulaire microvasculaire, un thème émergent dans le domaine des SVD. |
Notre équipe étudie les mécanismes physiopathogéniques des maladies des petits vaisseaux du cerveau (SVDs). Les SVDs sont responsables d’environ 25 à 30% des AVC ischémiques et d’environ 90% des hémorragies cérébrales, et sont une cause majeure de déclin cognitif et de handicap chez l’adulte; elles se manifestent à l’IRM cérébrale par des lésions étendues de la substance blanche et des infarctus lacunaires ou des hémorragies. Les SVDs sont sous tendues par des lésions dégénératives des petits vaisseaux du cerveau affectant leur structure ou leur fonction ; elles résultent d’une association complexe de facteurs génétiques et environnementaux, parmi lesquels l'hypertension artérielle et l'âge sont actuellement considérés comme les plus importants. Malgré l'importance des SVDs, leurs mécanismes physiopathogéniques sont très mal connus et il n'existe aucun traitement spécifique.
Les petits vaisseaux du cerveau ont des propriétés fonctionnelles uniques qui aident à s'assurer que le cerveau, qui a une réserve énergétique limitée, reçoive un apport suffisant de sang dans diverses conditions. Dans le cerveau, une augmentation de l’activité neuronale s'accompagne d'une augmentation du débit sanguin cérébral local (CBF) qui sert à satisfaire la demande accrue en glucose et en oxygène, un phénomène connu sous le nom d'hyperémie fonctionnelle ou de couplage neurovasculaire. Par ailleurs, les artères cérébrales et les artérioles se contractent ou se dilatent lorsque la pression intravasculaire augmente ou diminue, respectivement. Ce phénomène, connu sous le nom de réactivité myogénique, est un contributeur majeur du maintien du flux sanguin cérébral (autorégulation) au cours des variations physiologiques de la pression artérielle systémique.
La maladie CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) est un archétype de forme ischémique SVD, qui partage de nombreuses similitudes cliniques et pathologiques avec les formes sporadiques de SVDs. IL s’agit de la cause héréditaire la plus fréquente de démence vasculaire.
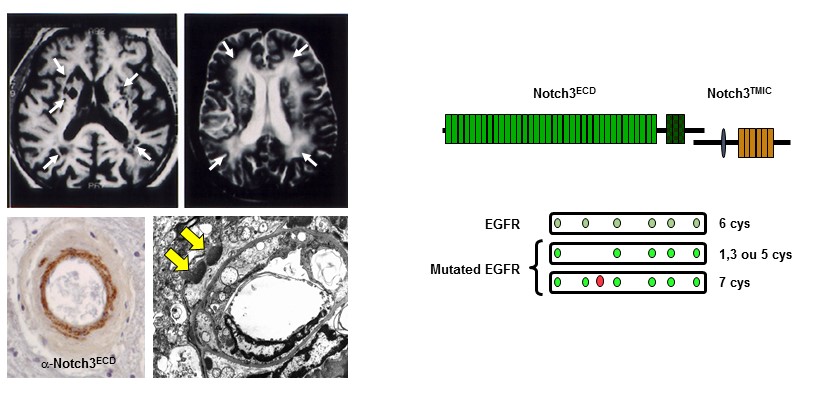
Figure 1: Caractéristiques (IRM, histologiques et moléculaires) cardinales de la maladie CADASIL
Notre groupe a démontré que CADASIL est causé par des mutations extrêmement stéréotypées qui modifient le nombre de résidus de cystéine dans le domaine extracellulaire de Notch3 (Notch3ECD), un récepteur principalement exprimé dans les cellules musculaires lisses vasculaires (CML), qui est essentiel pour la maturation et la fonction des petits vaisseaux cérébraux. Nous avons établi qu'une caractéristique pathologique centrale dans la maladie CADASIL est l'accumulation anormale du Notch3ECD dans les petits vaisseaux, dans des dépôts extracellulaires appelés GOM (Granular Osmiophilic Material). Nous avons généré une collection unique de modèles souris, dont un qui récapitule les principales manifestations de la maladie, et fait des contributions importantes dans la compréhension des mécanismes de la maladie. En particulier, nous avons montré que le couplage neurovasculaire, l’autorégulation du débit sanguin cérébral, le tonus myogénique et la dilatation maximale des artères cérébrales sont très précocement altérés dans le modèle CADASIL TgNotch3R169C.
La maladie COL4A1 / COL4A2, causée par des mutations dominantes dans la région codante des gènes COL4A1 ou COL4A2, est un archétype de forme hémorragique de SVD. Les gènes COL4A1 et COL4A2, qui codent respectivement pour les chaînes α1 et α2 du collagène de type IV, forment un hétérotrimère qui est un composant majeur des membranes basales vasculaires. Notre compréhension des mécanismes de cette maladie reste très rudimentaire et la prise en charge clinique des porteurs de mutations reste extrêmement difficile en raison de la pénétrance incomplète des hémorragies et de leur expressivité variable allant de microsaignements asymptomatiques à des hémorragies cérébrales menaçant le pronostic vital.
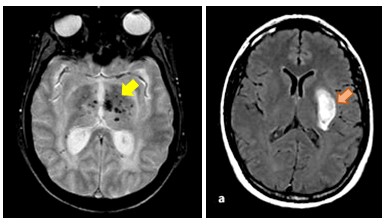
Figure 2: Microsaignements (flèche jaune) et hémorragie intracérébrale (flèche orange) dans la COL4A1/ COL4A2
Notre but ultime est de déchiffrer les mécanismes pathogènes des SVDs et de développer des thérapies efficaces pour ces maladies. Notre concept général est que les SVDs monogéniques - en particulier les maladies CADASIL et COL4A1 - offrent une opportunité unique pour identifier ces mécanismes et développer des stratégies thérapeutiques qui peuvent être validées dans des modèles animaux prédictifs.
Articles principaux: Fouillade et al., Arterioscler Thromb Vasc Biol. 2013; Monet-Leprêtre M et al, Brain. 2013; Cognat et al, Stroke 2014; Dabertrand et coll., Proc Natl Acad Sci U S A. 2015; Capone et al, Ann Neurol., 2016; Capone et al, eLife 2017; Baron-Menguy et al., Hypertension, 2017
Brevet : WO / 2016/04653 "Traitement immunologique de la maladie CADASIL"
- Bien que quelques mutations Notch3 abolissent ou réduisent de façon incontestable l’activité du récepteur Notch3 muté, nous avons obtenu des preuves convaincantes que les manifestations cérébrovasculaires dans la maladie CADASIL ne sont pas le résultat d'une réduction de l'activité de Notch3, du moins au stade précoce de la maladie. Au lieu de cela, nous avons démontré que la mutation Arg169Cys Notch3, une mutation récurrente archétypale, entraine une augmentation de l’activité de Notch3 qui est impliquée dans la réduction de la dilatation maximale des artères cérébrales.
- Nous avons établi que les mutations CADASIL conduisent à un processus complexe d'agrégation de Notch3ECD en espèces multimériques qui se complexent avec des protéines associées aux protéines de la matrice extracellulaire (ECM), telles que TIMP3 et la vitronectine, pour former les GOM. En conduisant des études d'interactions génétiques avec le modèle murin TgNotch3R169C, nous avons prouvé que l’accumulation anormale de TIMP3 et de la vitronectine était impliquée dans le processus pathogène.
- Nous avons identifié une nouvelle voie de signalisation, ADAM17 / HB-EGF / (ErbB1 / ErbB4)/ KV1, régulée négativement par TIMP3, qui est essentielle dans la régulation du tonus myogénique des artères cérébrales et des réponses évoquées du débit sanguin cérébral. Nous avons ensuite démontré que l’inhibition de cette voie de signalisation du fait de l’excès de TIMP3 était impliquée dans la dysfonction des vaisseaux cérébraux dans le modèle souris CADASIL.

Figure 3: Modèle illustrant le mécanisme par lequel les mutations Notch3 altèrent la fonction des vaisseaux cérébraux dans la maladie CADASIL
- Au sein de notre collection d'anticorps monoclonaux dirigés contre Notch3ECD, nous avons découvert qu'un anticorps administré par voie périphérique se lie spécifiquement aux dépôts de Notch3ECD dans les vaisseaux périphériques et cérébraux, malgré l'absence de fuite de la barrière hémato-encéphalique. Dans une étude préventive de type preuve de concept, nous avons établi que l’administration chronique de cet anticorps protégeait les souris CADASIL TgNotch3R169C des lésions vasculaires.
Articles principaux: Joutel et al. J Cereb Blood Flow Metab.2016; Ratelade et al, J. Pathol sous presse
- Nous avons montré que les souris knock-in exprimant la mutation pathogène p.Gly498Val dans la région amino-terminale de COL4A1 (générées dans le laboratoire d'E. Plaisier, Inserm U1151), récapitulent fidèlement la maladie humaine, avec en particulier la survenue de microsaignements, dont la pénétrance est complète, et de macrohémorragies dont la pénétrance est incomplète.
- Nous avons prouvé que les mécanismes mis en jeu dans les microsaignements et les macrohémorragies sont différents : les microsaignements résultent d’une fuite transitoire des capillaires au début de la vie postnatale, les macrohémorragies résultent d’une dégénérescence focale et segmentaire des cellules musculaires lisses au niveau des artères cérébrales apparaissant avec l’âge. Nous avons en outre montré que les lésions artérielles peuvent être évaluées au niveau de la rétine et nous avons prouvé que leur sévérité était fortement corrélée à la survenue des macrohémorragies, offrant la perspective d'identifier les patients à haut risque par un examen des artères de la rétine.
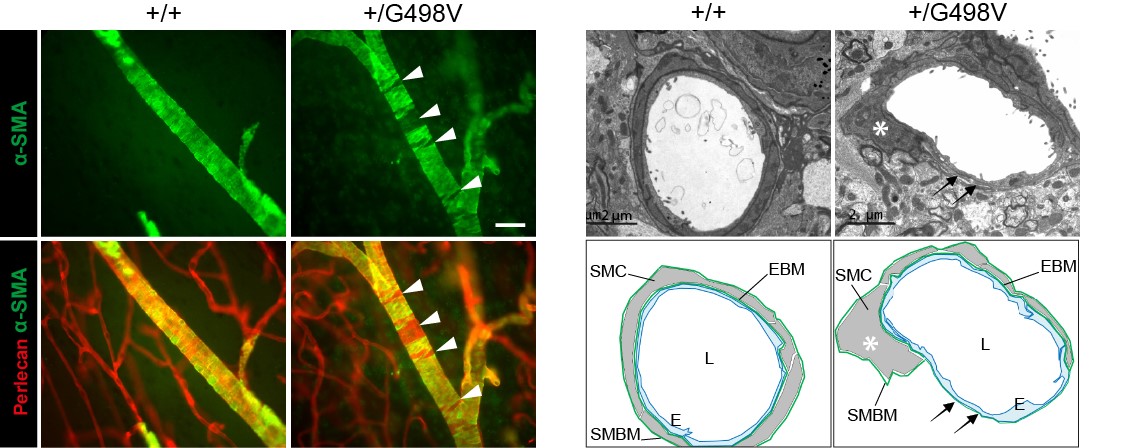
Figure 4: Défauts des cellules musculaires lisses artérielles chez les souris mutantes Col4a1
Ci dessous vous pouvez télécharger nos principaux articles ou résumés
 Reducing Hypermuscularization of the Transitional Segment between Arterioles and Capillaries Protects Against Spontaneous Intracerebral Hemorrhage.
Reducing Hypermuscularization of the Transitional Segment between Arterioles and Capillaries Protects Against Spontaneous Intracerebral Hemorrhage.
 Notch3ECD Immunotherapy Improves Cerebrovascular Responses in CADASIL Mice
Notch3ECD Immunotherapy Improves Cerebrovascular Responses in CADASIL Mice
 Mechanistic insights into a TIMP3- sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics
Mechanistic insights into a TIMP3- sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics
 Reducing Timp3 or Vitronectin Ameliorates Disease Manifestations in CADASIL Mice
Reducing Timp3 or Vitronectin Ameliorates Disease Manifestations in CADASIL Mice
 Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease
Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease
JRS - Accident vasculaire cérébral : de la clinique à la physiopathologie : A. Joutel
 Caractéristiques (IRM, histologiques et moléculaires) cardinales de la maladie CADASIL
Caractéristiques (IRM, histologiques et moléculaires) cardinales de la maladie CADASIL
 Microsaignements (flèche jaune) et hémorragie intracérébrale (flèche orange) dans la COL4A1/ COL4A2
Microsaignements (flèche jaune) et hémorragie intracérébrale (flèche orange) dans la COL4A1/ COL4A2
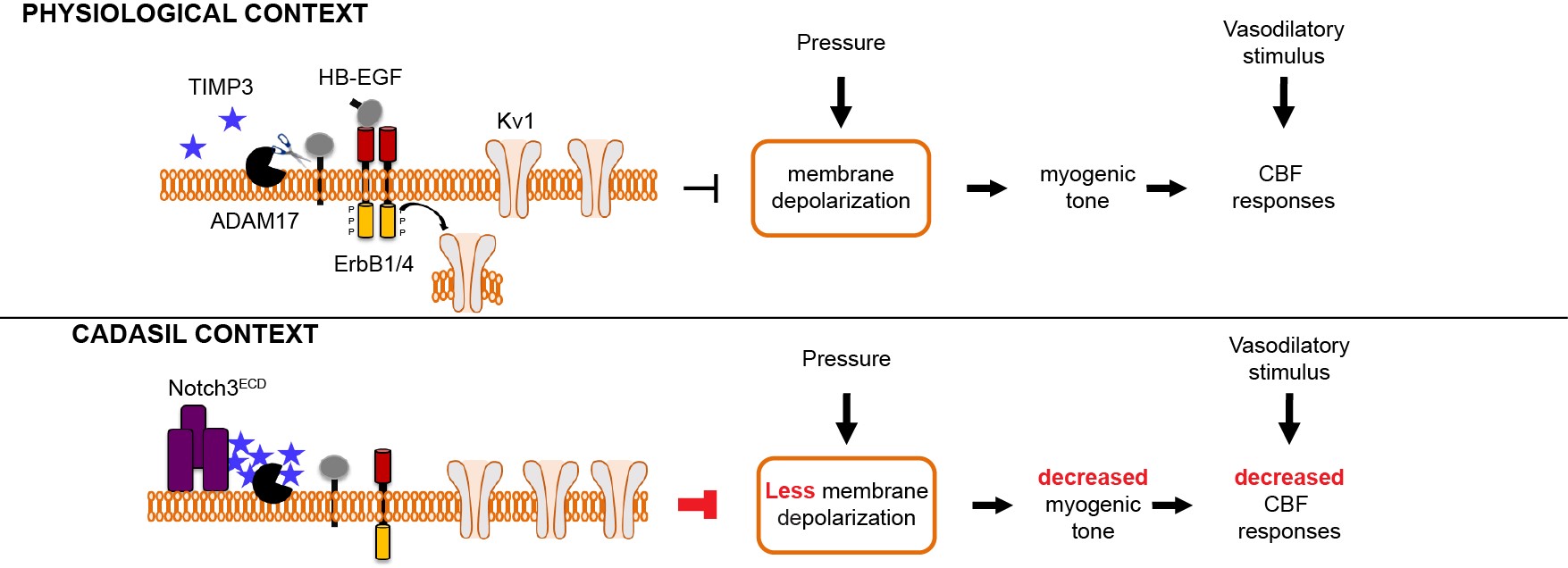 Modèle illustrant le mécanisme par lequel les mutations Notch3 altèrent la fonction des vaisseaux cérébraux dans la maladie CADASIL
Modèle illustrant le mécanisme par lequel les mutations Notch3 altèrent la fonction des vaisseaux cérébraux dans la maladie CADASIL
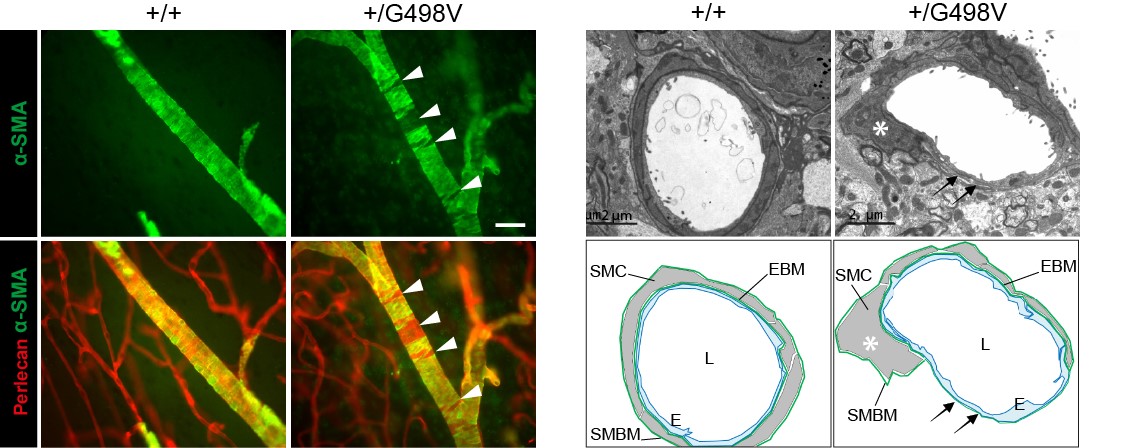 SMC defects in Col4a1 mutant mice
SMC defects in Col4a1 mutant mice
Fondation Leducq: (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain)
![]()
ANR (programme RHU “From Target Identification to Next Generation Therapies for cerebral Small Vessel Diseases”)

Union Européenne (Horizon 2020 Programme de recherches et d'innovations)

Fondation Maladies Rares




