Team leader : Anne Joutel
Team member : Anaele Arnou | Shiraz DIB | Valérie Domenga-Denier | Antoine Drieu | Nicolas Dupré | Julien Guemri | Florian Gueniot | Sherlina Salignac
 |
Cerebral small vessel diseases (SVD) account for ~25% of ischemic strokes, ~ 90% of intracerebral hemorrhages (ICH) and are a leading cause of cognitive decline and disability. Our limited understanding of the pathogenesis of SVD is a major obstacle to the development of treatments. Monogenic forms of SVD, such as CADASIL, an ischemic form of SVD caused by highly stereotyped mutations in the Notch3 receptor, and COL4A1/2-related ICH, caused by mutations in the alpha1 or alpha2 chains of collagen type IV, provide a window into the mechanism underlying much more common, but largely indistinguishable, sporadic forms of SVD. Over the past years, our lab has generated and characterized several mouse models of CADASIL and COL4A1/2-related ICH. In CADASIL, we established that aggregation/accumulation of the extracellular domain of Notch3 in brain vessels is a central event, promoting the abnormal recruitment of functionally important extracellular matrix proteins that ultimately cause multifactorial toxicity. We identified a novel mechanism linking pathological alterations of the vascular extracellular matrix and a channelopathy-like defect that underlies cerebrovascular dysfunction. Moreover, we obtained preliminary evidence of efficacy of passive immunization targeting Notch3 in a proof-of-concept study. For the next years, we aim to elucidate the mechanism of lacunar infarcts, to identify which Notch3 species are toxic and to further investigate the therapeutic potential of passive immunization against Notch3, with the goal to advance it towards clinical trial readiness. In COL4A1-related ICH, we recently identified the cellular mechanisms of ICH and we now aim to decipher the molecular mechanisms. Finally, we have ambition to move on to the study of sporadic SVD, starting with the analysis of the microvascular ECM, an emerging theme in the field of SVD. |
Our group studies the pathogenesis of small vessel diseases (SVDs) of the brain, which refer to pathological processes that affect the structure and function of small penetrating vessels within the brain. SVDs account for ~25-30% of ischemic strokes, ~ 90% of cerebral hemorrhages and are a leading cause of cognitive decline and disability. The salient consequences of SVD are lacunar infarcts or hemorrhages in the white and/or deep gray matter, and the development of widespread white matter lesions. SVDs appear driven by a complex mix of genetic and environmental factors, among which age and arterial hypertension are currently deemed the most important. Despite the importance of SVDs, pathogenesis remains obscure and there are currently no specific treatments.
Small vessels of the brain have unique functional properties that help ensure that the brain, which has a limited fuel reserve, receives an adequate supply of blood under a variety of conditions. In the brain, increased cellular activity is accompanied by an increase in local cerebral blood flow (CBF) that serves to satisfy enhanced glucose and oxygen demand, a phenomenon known as functional hyperemia or neurovascular coupling. Cerebral arteries and arterioles also constrict or dilate when intravascular pressure increases or decreases, respectively. This phenomenon, known as myogenic reactivity, is a major contributor to the maintenance of cerebral blood flow (autoregulation) during variations in systemic blood pressure.
CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) is an archetypal ischemic form of SVD, which share a number of clinical and pathological features with sporadic SVDs. Notably, CADASIL emerges as the most common heritable cause of vascular dementia worldwide.
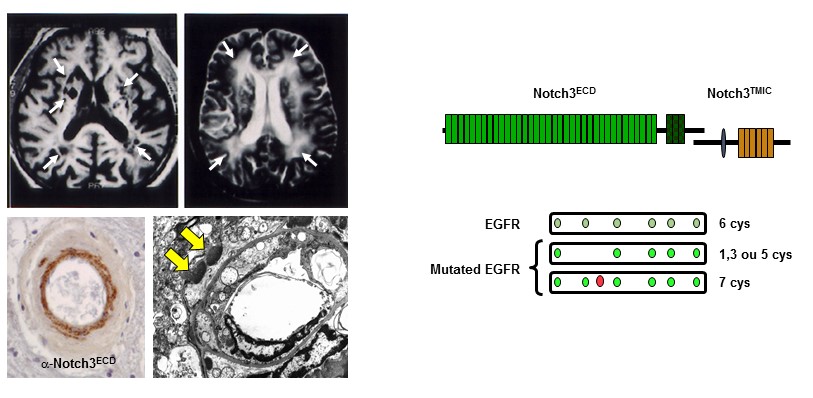
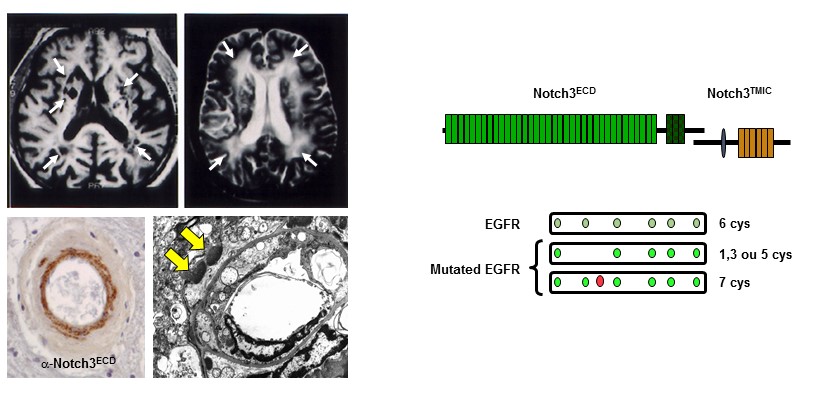
Figure 1: Neuroimaging, vascular and molecular features of CADASIL
Over the past years, our group has produced some of the seminal work in this field. We demonstrated that CADASIL is caused by highly stereotyped mutations that alter the number of cysteine residues in the extracellular domain of NOTCH3 (Notch3ECD), a receptor that is predominantly expressed in vascular smooth muscle cells (SMCs) and is critical for the maturation and function of small brain vessels. We established that one central pathological feature is the abnormal accumulation of Notch3ECD in small vessels, in extracellular deposits called Granular Osmiophilic Material (GOM). We have generated a unique collection of mouse models, including one that recapitulates the salient features of the disease, and made major progress in identifying disease mechanisms. We showed that cerebrovascular dysfunction, with impaired neurovascular coupling and CBF autoregulation, inward remodeling (reduced maximal dilation) and decreased myogenic tone of cerebral arteries are early and prominent features in the well-established TgNotch3R169C CADASIL model.
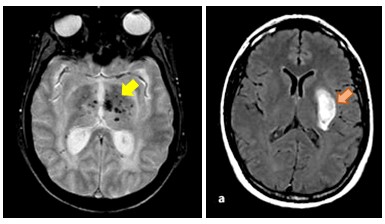
COL4A1/COL4A2-related SVD, caused by dominant mutations in the coding region of COL4A1 or COL4A2, is an archetypal form of hemorrhagic SVD. COL4A1 and COL4A2 genes, which encode collagen type IV α1 and α2 chains respectively, form a heterotrimer that is a major component of vascular basement membranes. Yet, our understanding of the pathogenesis is still limited and clinical management of mutation carriers remains extremely challenging owing to the partial penetrance of hemorrhages and their variable expressivity that range from asymptomatic microhemorrhage to life-threatening macrohemorrhage.
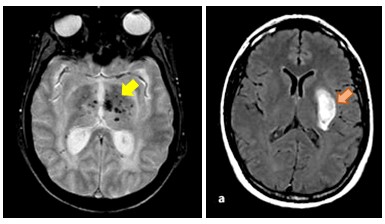
Figure 2: Microbleeds and intracerebral hemorrhage in COL4A1-related SVD
Our ultimate goal is to decipher the pathogenic mechanisms of SVDs and develop effective therapies for these diseases. Our overarching concept is that genetic SVDs – especially CADASIL and COL4A1/COL4A2-related SVD– provide a unique opportunity to identify these mechanisms and develop therapeutic strategies that can be validated in predictive animal models.
Main papers: Fouillade et al, Arterioscler Thromb Vasc Biol. 2013; Monet-Leprêtre M et al, Brain. 2013; Cognat et al, Stroke 2014; Dabertrand et al, Proc Natl Acad Sci U S A. 2015 ; Capone et al, Ann Neurol., 2016 ; Capone et al, eLife 2017; Baron-Menguy et al, Hypertension, 2017
Patent WO/2016/04653: “Immunological treatment of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy »
- Although a few CADASIL-associated NOTCH3 mutations unambiguously result in abolished or reduced Notch3 activity, we obtained compelling evidence that cerebrovascular manifestations are not the result of a reduction in Notch3 activity. Instead, we demonstrated that the archetypal Arg169Cys Notch3 mutation is associated with increased Notch3 activity to cause inward remodeling of cerebral arteries.
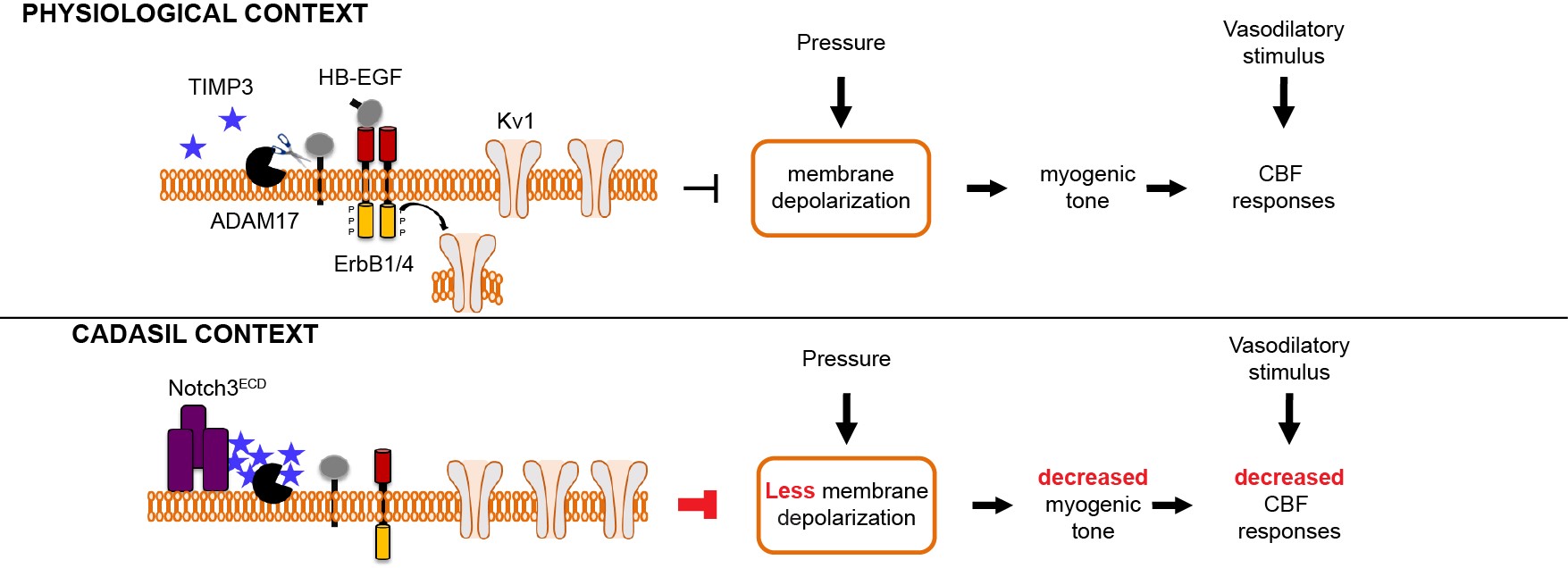
- We found that CADASIL mutations lead to a complex process of Notch3ECD aggregation into multimeric species that complex with proteins associated with the extracellular matrix (ECM) proteins, including, but not limited to, TIMP3 and vitronectin, and eventually form granular osmiophilic material (GOM) deposits. Using genetic interaction studies in the TgNotch3R169C mouse model, we established that elevated levels of TIMP3 and vitronectin, acting downstream of Notch3ECD deposition, play a role in cerebrovascular manifestations of CADASIL.
- We identified ADAM17/HB-EGF/(ErbB1/ErbB4)/KV1 as a key TIMP3-sensitive signaling module that is essential for maintaining robust CBF responses to evoked neural activity as well as for myogenic responses of brain arteries. Next, we provided evidence that elevated TIMP3 blunts the ADAM17/HB-EGF/(ErbB1/ErbB4)/KV1 pathway to reduce myogenic tone and cause CBF deficits.
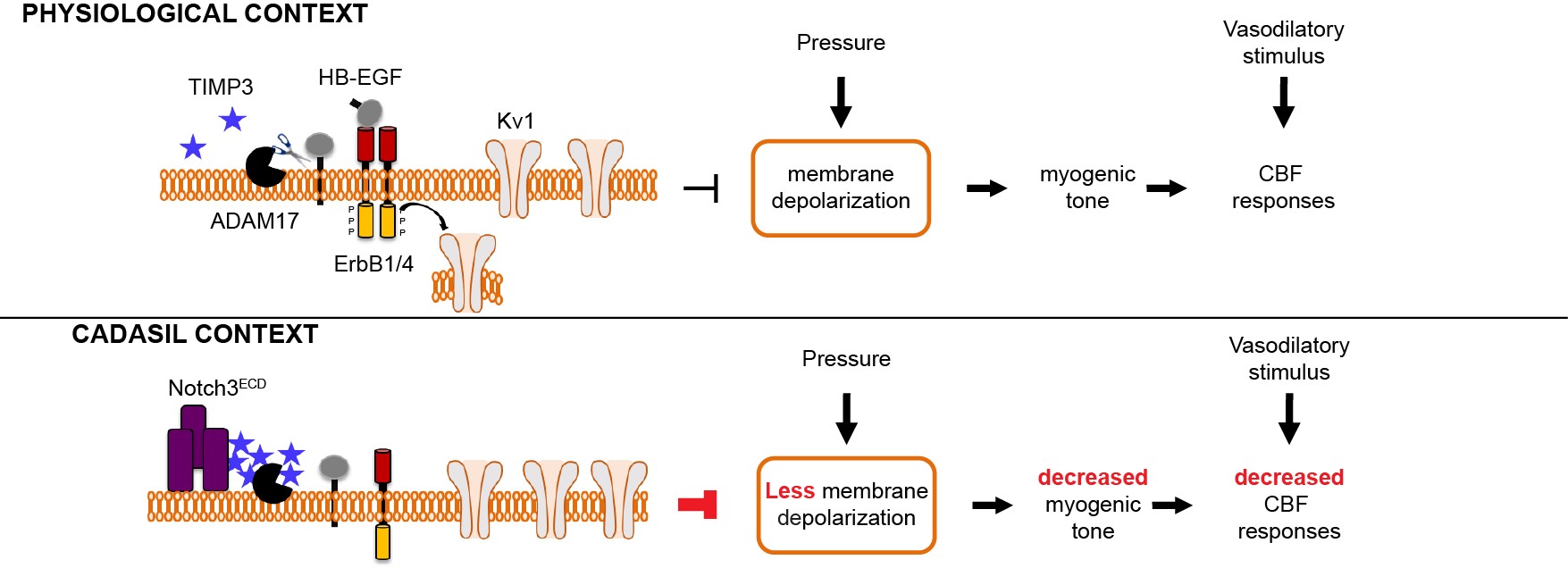
Figure 3: Working model of how Notch3 mutation decreases myogenic tone to cause CBF deficits
- Within our collection of monoclonal antibodies raised against Notch3ECD, we found that one peripherally-administered antibody specifically binds to Notch3ECD deposits in both the peripheral and cerebral vasculature, despite the absence of blood brain barrier leakage. Further, in a proof-of-concept preventive study, we found that TgNotch3R169C mice treated with this antibody were fully protected against inward remodeling, reduced myogenic tone and CBF deficits.
Main papers: Joutel et al, J Cereb Blood Flow Metab.2016; Ratelade et al, J. Pathol in press
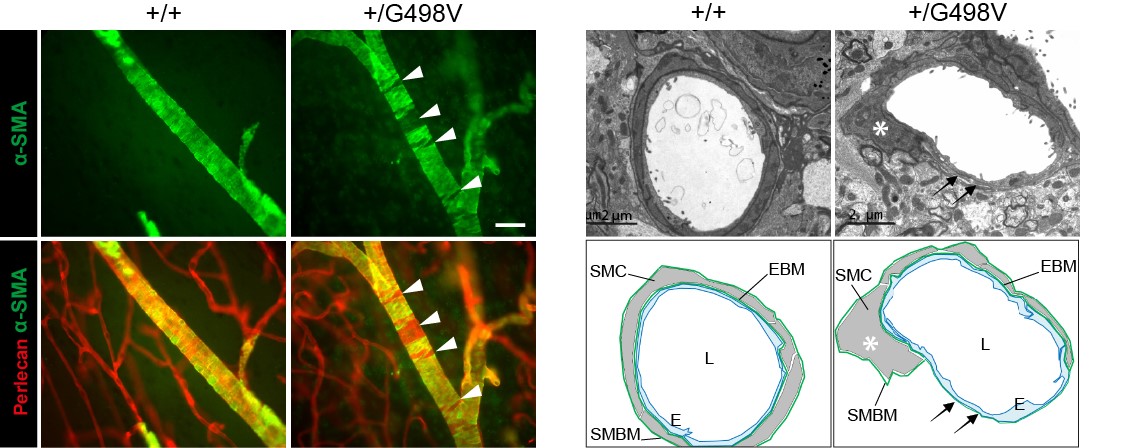
- We found that knock-in mice harboring the p.Gly498Val disease-causing mutation in the amino-terminus half of COL4A1 (generated in E. Plaisier’s lab, Inserm U1151), faithfully recapitulate the human disease, with highly penetrant microhemorrhages and macrohemorrhages with reduced penetrance.
- We provided evidence that microhemorrhages and macrohemorrhages are driven by two distinct mechanisms, transient leakage of capillaries in the early postnatal life for microhemorrhages and age-related smooth muscle cell degeneration in a subset of deep arteries for macrohemorrhages. We further showed that arterial lesions can be assessed in the retina and that inter-individual variability in their severity is a critical factor underlying the partial penetrance of macrohemorrhage, raising the prospect of identifying the high-risk patients.
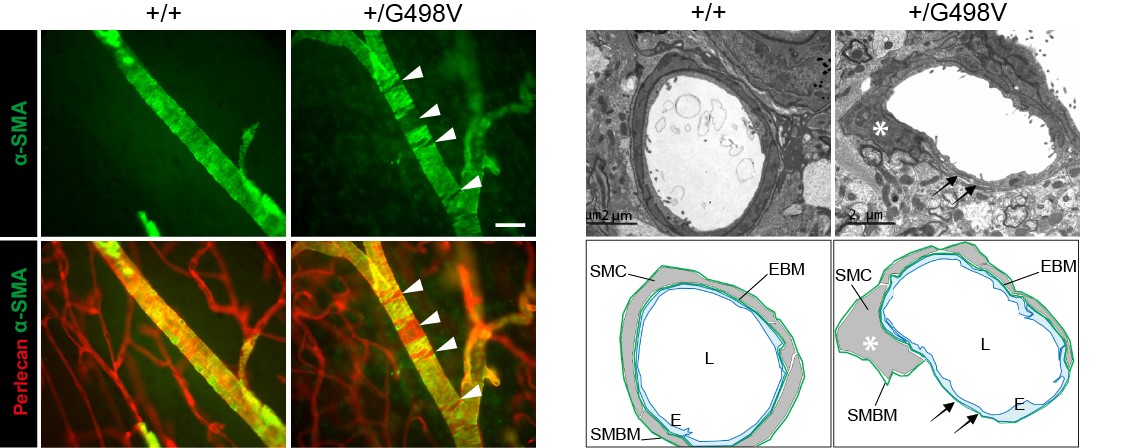
Figure 4: SMC defects in Col4a1 mutant mice
Below you can download the main publications or abstracts
 Reducing Hypermuscularization of the Transitional Segment between Arterioles and Capillaries Protects Against Spontaneous Intracerebral Hemorrhage.
Reducing Hypermuscularization of the Transitional Segment between Arterioles and Capillaries Protects Against Spontaneous Intracerebral Hemorrhage.
 Notch3ECD Immunotherapy Improves Cerebrovascular Responses in CADASIL Mice
Notch3ECD Immunotherapy Improves Cerebrovascular Responses in CADASIL Mice
 Mechanistic insights into a TIMP3- sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics
Mechanistic insights into a TIMP3- sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics
 Reducing Timp3 or Vitronectin Ameliorates Disease Manifestations in CADASIL Mice
Reducing Timp3 or Vitronectin Ameliorates Disease Manifestations in CADASIL Mice
 Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease
Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease
JRS - Stroke: from clinic to physiopathology: A. Joutel
Fondation Leducq: (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain)
![]()
ANR (programme RHU “From Target Identification to Next Generation Therapies for cerebral Small Vessel Diseases”)

European Union (Horizon 2020 Research and Innovation Programme)

Rare Diseases Foundation